Quickstart¶
Time: 10 minutes
preqc-lr generates a PDF report containing several plots such as estimated genome size and coverage. This report can be used to evaluate the quality of your sequencing data. Here, we provide a step-by-step tutorial to get you started!
Requirements:
Download example dataset¶
You can download the example dataset we will use here:
wget http://s3.climb.ac.uk/nanopolish_tutorial/preqclr_example_data.tar.gz
tar -xf preqclr_example_data.tar.gz
cd example_data/
Details:
This dataset from an E. coli sample were produced using Oxford Nanopore Technologies (ONT) MinION sequencer.
- Sample : E. coli str. K-12 substr. MG1655
- Instrument : ONT MinION sequencing R9.4 chemistry
- Basecaller : Albacore v2.0.1
- Number of reads: 63931
Generate overlap information with minimap2¶
We use minimap2 to find overlaps between our ONT long reads:
minimap2 -x ava-ont albacore_v2.0.1-merged.fasta albacore_v2.0.1-merged.fasta > overlaps.paf
If we take a peek at the first few lines of the Pairwise mApping Format (PAF) file, we see the following:
7fd051aa-c88b-4cf7-8846-cc2117780be2_Basecall_1D_template 6605 118 6425 - ae8fc44b-ee05-4c7a-a611-483bb408cb9e_Basecall_1D_template 7834 629 7230 24806671 0 tp:A:S cm:i:387 s1:i:2413 dv:f:0.1144
7fd051aa-c88b-4cf7-8846-cc2117780be2_Basecall_1D_template 6605 343 6417 - cecc6ee9-f1ec-4c82-915a-5312f39f7ec5_Basecall_1D_template 6762 421 6710 24286372 0 tp:A:S cm:i:370 s1:i:2374 dv:f:0.1149
7fd051aa-c88b-4cf7-8846-cc2117780be2_Basecall_1D_template 6605 118 6377 - c0d8087f-ad9f-430c-8094-24c6187bed6c_Basecall_1D_template 11415 3039 9493 22646559 0 tp:A:S cm:i:346 s1:i:2209 dv:f:0.1214
7fd051aa-c88b-4cf7-8846-cc2117780be2_Basecall_1D_template 6605 738 6422 - bbb93738-16ec-4bcd-86e5-31e852946a7d_Basecall_1D_template 6596 553 6498 20916000 0 tp:A:S cm:i:302 s1:i:2031 dv:f:0.1242
7fd051aa-c88b-4cf7-8846-cc2117780be2_Basecall_1D_template 6605 212 6422 - 943b8d89-2ee5-4d67-91d1-a94772afed31_Basecall_1D_template 7324 807 7152 20676448 0 tp:A:S cm:i:322 s1:i:2011 dv:f:0.1255
You can find more information about the format of the PAF file here.
Generate assembly graph with miniasm¶
We use miniasm to get an assembly graph in the Graphical Fragment Assembly format:
miniasm -f albacore_v2.0.1-merged.fasta overlaps.paf > layout.gfa
Note
Make sure layout.gfa and overlaps.paf are not empty before continuing.
Perform calculations¶
We now have the necessary files to run preqc-lr (albacore_v2.0.1-merged.fasta, overlaps.paf, and layout.gfa).
To generate the data needed for the report we first run preqc-lr-calculate
./preqclr \
--reads albacore_v2.0.1-merged.fasta \
--sample_name ecoli.ONT \
--paf overlaps.paf \
--gfa layout.gfa \
--verbose
This will produce a JSON formatted file (ecoli.ONT.preqclr) and a log of calculations that were performed (ecoli.ONT_preqclr-calculate.log).
Generate report¶
Now we are ready to run preqclr-report to generate a PDF file describing quality metrics of the sequencing data:
python preqclr-report.py \
-i ecoli.ONT.preqclr --verbose
This will produce a PDF file: ecoli.ONT.pdf.
Example report¶
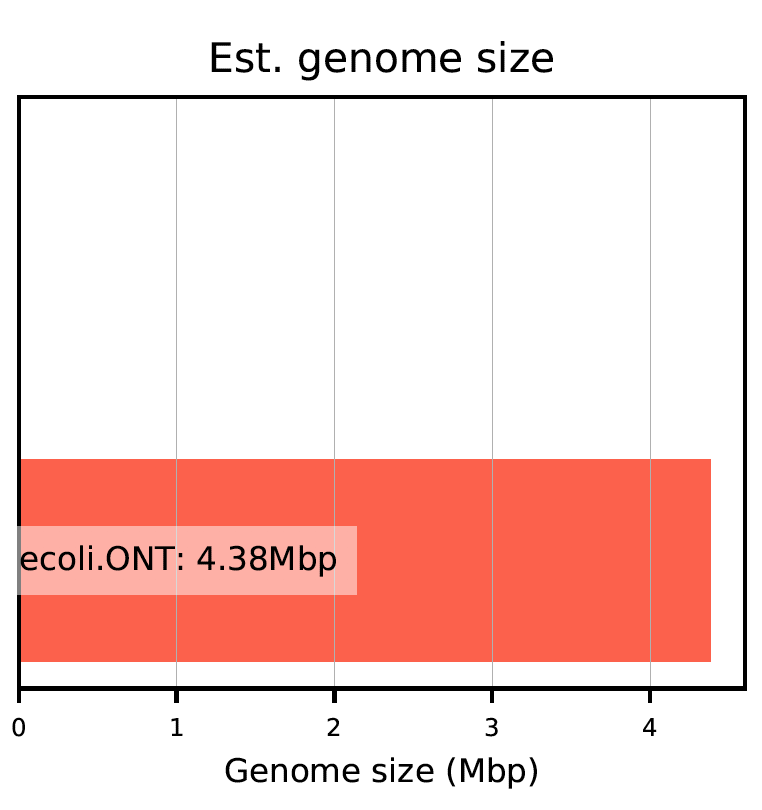
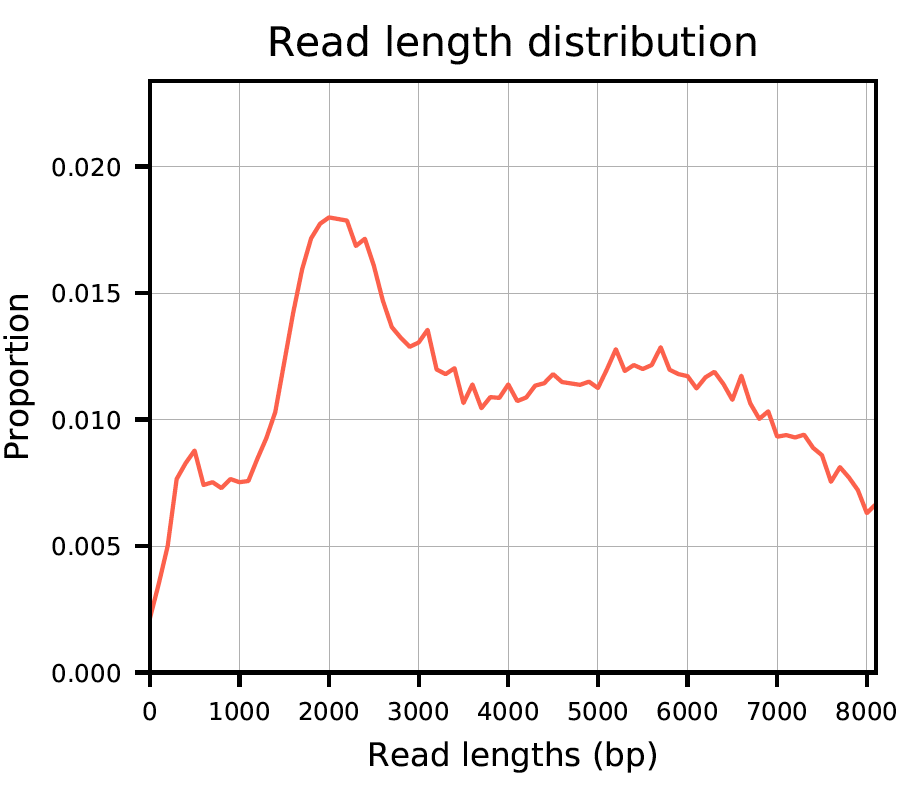
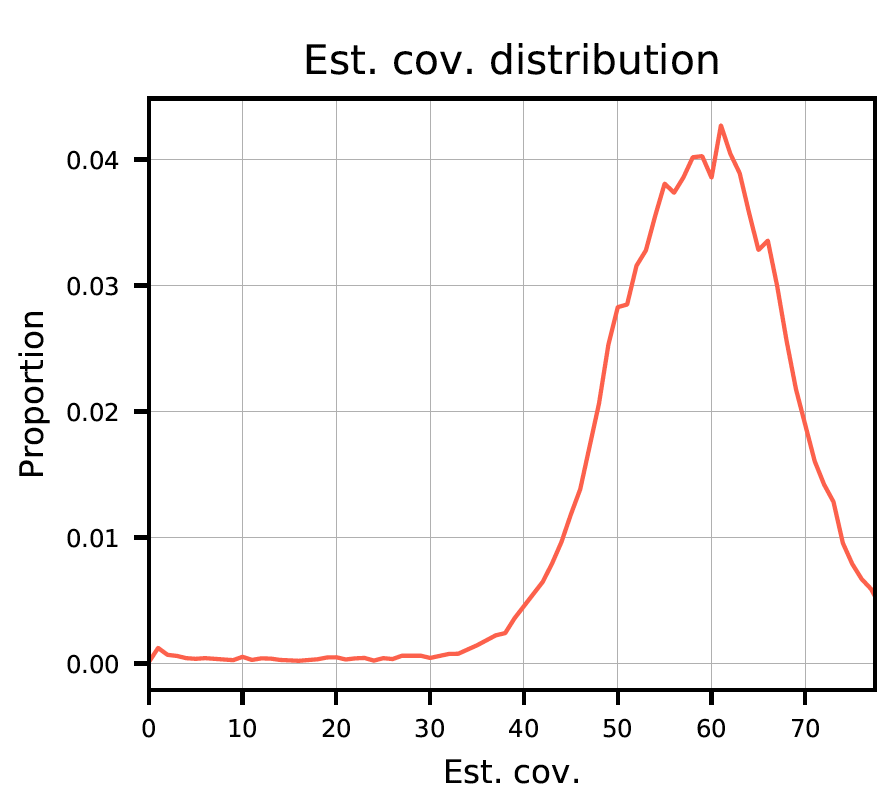
The report produces plots as seen below.
Plot 0:
Plot 1:
Plot 2:
Plot 3:
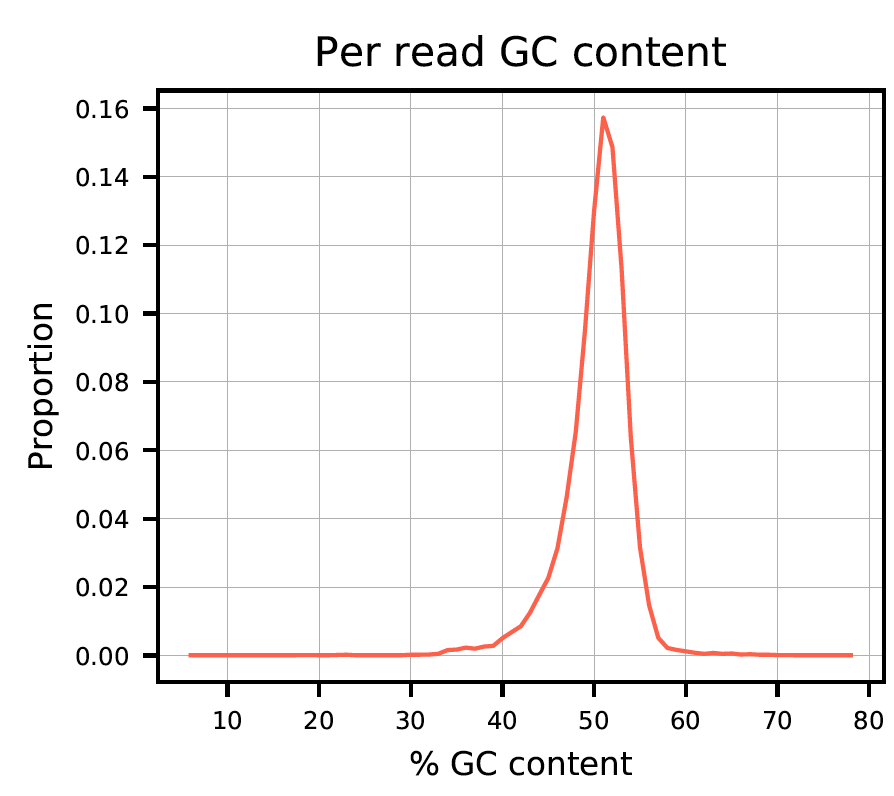
Plot 4:
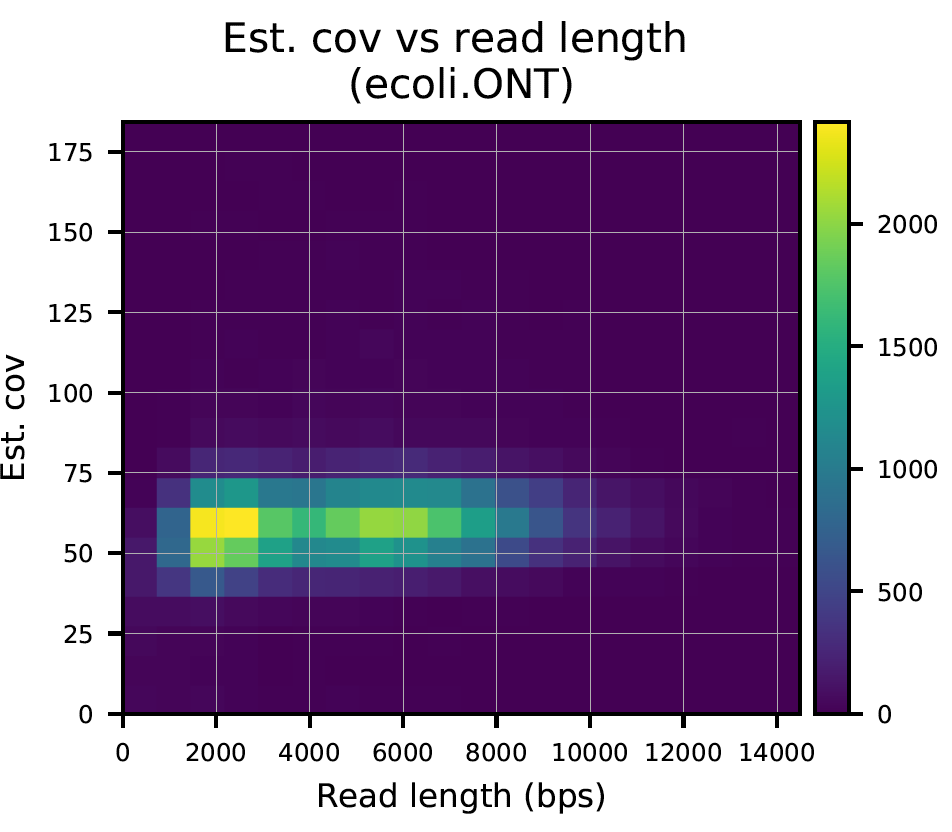
Plot 5:
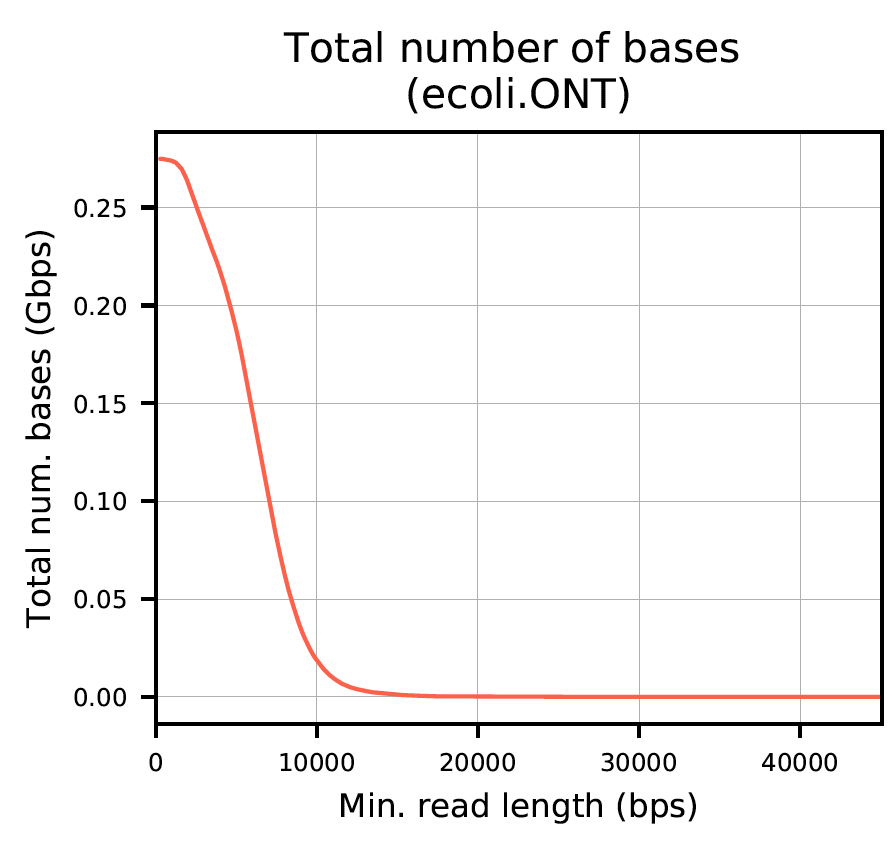
Plot 6: